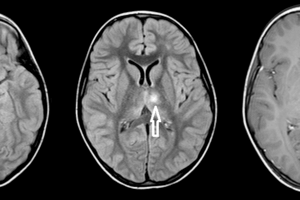
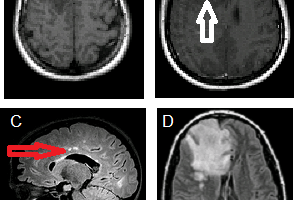
Neurosarcoidosis is defined as neuronal, either central or peripheral, involvement of sarcoidosis. About 10% of patients with systematic sarcoidosis have significant neurological deficits as a result of granulomatous lesions, but subclinical disease may be present in another 5% of patients with sarcoidosis.
Clinical Presentation of Neurosarcoidosis
Neurosarcoidosis can affect almost any part of the nervous system and therefore can have myriad presentations, making diagnosis challenging.
Cranial neuropathies
The most common presentations include cranial neuropathies (50-75% of neurosarcoid cases), with either a unilateral (65%) or bilateral (35%) cranial nerve 7 palsy being the most frequently affect (should be differentiated from Bell’s palsy or Ramsay Hunt Syndrome). The optic nerve (CN 2) is the next most commonly affected cranial nerve, and deficits can range from blurry vision to permanent loss of vision.
Heerfordt’s Syndrome is a classic presentation of neurosarcoid that includes a facial nerve palsy, parotid gland swelling, fever and uveitis.
Parenchymal brain lesions
About half of patients with neurosarcoidosis will have parynchemal brain lesions. White matter is often involved, and can appear very similar to the lesions of multiple sclerosis. However, neurosarcoidosis has tendency to affect the base of the brain, including pons, midbrain, the pituitary gland and hypothalamus. As a result, severe hormonal dysregulation often accompanies neurosarcoid with symptoms as varied as electrolytes and fluid imbalances, dysmenorrhea, galactorrhea, morbid obesity, insomnia, thermal dysregulation and personality changes.
Other presentations
Granulomatous tissue can also develop in the spinal cord producing weakness, upper motor lesions at specific cord levels, syrinx, etc. in about 10% of neurosarcoid patients.
Behavioral and cognitive issues develop in about 20% of patients.
Both peripheral nerves and muscles can be directly affected and produce symptoms that localize to those structures.
Pathology
Sarcoidoisis is an TH1 (type 1 T helper) cell mediate autoimmune disorder in which non-caseating granulomas develop. Both genetic and environmental factors contribute to the development and severity of disease. In particular, exposure to molds, metal powders, and chemicals or particles encountered in the firefighting career increase the risk of developing non-caseating granulomas. M tuberculosis and P acnes infection also increase the risk of sarcoidosis.
Diagnosis
MRI with gadoinium is currently the optimal imaging modality for detecting neurosarcoidosis because the granulomatous infiltrates enhance. Typical findings including white matter lesions and leptomeningeal enhancement.
Lumbar puncture findings include elevated protein, mononuclear cell pleocytosis, a CD4 to CD8 ratio greater than 5, oligoclonal bands, and elevated angiotensin converting enzyme.
Tissue biopsy is usually definitive since non-caseating granulomas are characteristic of sarcoidosis. Other CNS disorders that can have an MRI appearance similar to neurosarcoid, CNS lymphoma for example, will not have granulomas.
Epidemiology
While sarcoidosis can be found world wide, it has a higher prevelance in Americans of West African descent and in North European Caucasions than in other populations.
The age of onset varies by ethnicity. In those of African descent the onset is typically in the 3rd and 4th decades (20s and 30s), but in women of Scandinavian or Japanese descent there is a second peak in the 6th decade of life and older (older than 50).
In the US, sarcoidosis has a prevalence of 40 per 100,000 (so about 4 per 100,000 have neuroscaroidosis).
Management
A definitive diagnosis of neurosarcoid opens up several therapeutic approaches.
First line therapy consists of corticosteroids, typically prednisone, with or without hydroxychloroquine, pentoxifylline, or thalidomide.
Second line therapy severe symptoms includes infliximab, which acts as a TNF-alfpha blocker or cyclophosphamide. For cases in which symptoms are mild to moderate but for which prednisone has failed, second line therapies include mycophenolate mofetil, methotrexate, azathioprine, or hydroxychloroqine.
References
Neurosarcoidosis: presentations and management. Terushkin V, Stern BJ, Judson MA, Hagiwara M, Pramanik B, Sanchez M, Prystowsky S. Neurologist. 2010 Jan;16(1):2-15. Review. Erratum in: Neurologist. 2010 Mar;16(2):140.