Pompe disease is an autosomal recessive lysosomal glycogen storage disease. Mutations in the acid alpha-glucosidase (GAA) gene on chromosome 17q25.2-q25.3 result in abnormal accumulation of glycogen within lysosomes and subsequent lysosomal dysfunction.
Types of Pompe disease
Infantile Pompe’s disease
Infantile Pompe’s disease presents in the first months of life with failure to thrive, muscle weakness that manifests as floppiness and head lag. Untreated the disease progress includes cardiomegaly, enlarged tongues, and respiratory difficulties with frequent lung infections that can proceed to respiratory failure. Before the development of recombinant acid alpha-glucosidase most babies would die before their first birthday.
Non-infantile Pompe’s disease
Late-onset Pompe’s disease (also known as Smith disease for late infantile presentation or Engel disease for the adult form of acid apha-glucosidase deficiency) has a slower progression. In one series of patients first symptoms were noted anywhere from five years old to 45 years old (Hagemans et al., 2005). Difficulty running was typically the first symptom noticed. Age at diagnosis ranged from 18 to 50.
Pathological findings
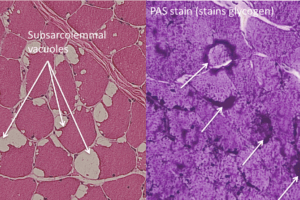
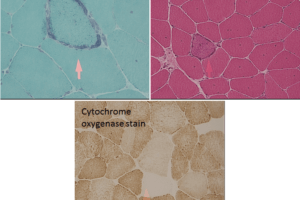
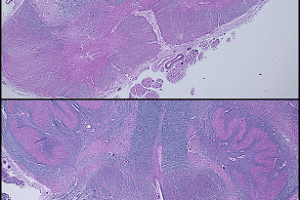
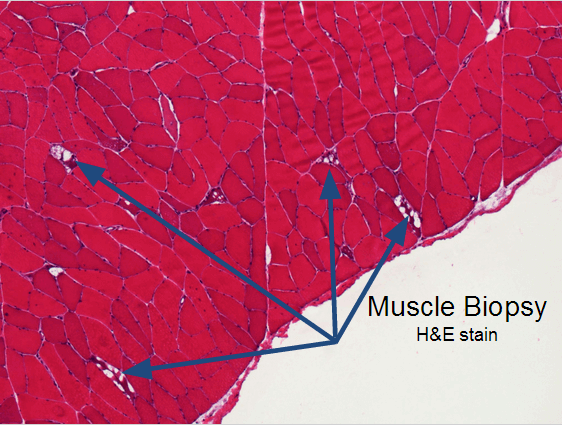
These slides are from an adult case of Pompe’s disease, so the findings are less severe than would be found in an infantile case.
Classic findings on light microscopy include:
Vacuolar myopathy.
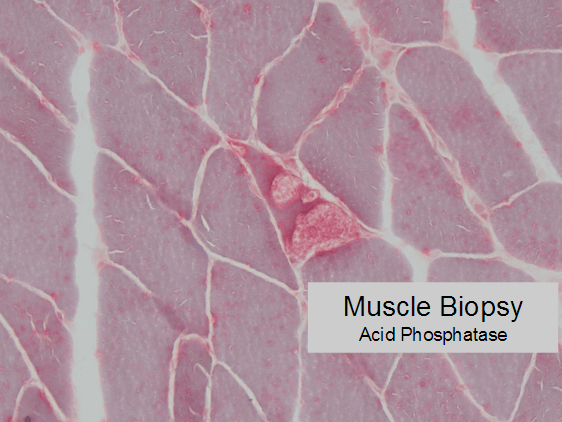
The vacuoles stain positive for acid phosphatase activity, suggesting they are lysosomes.
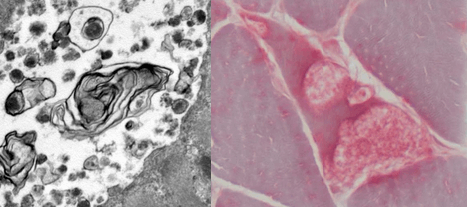
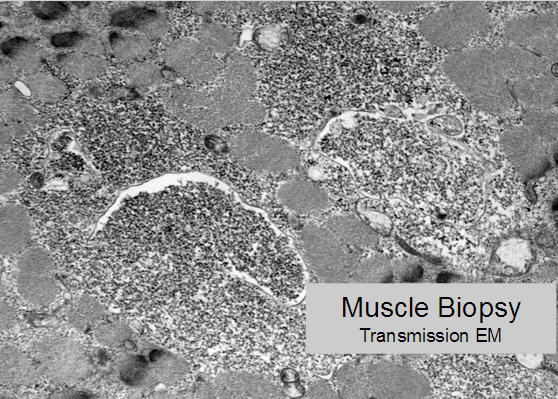
Glycogen accumulates within lysosomes (membrane-bound granular material) visible on transmission electron microscopy.
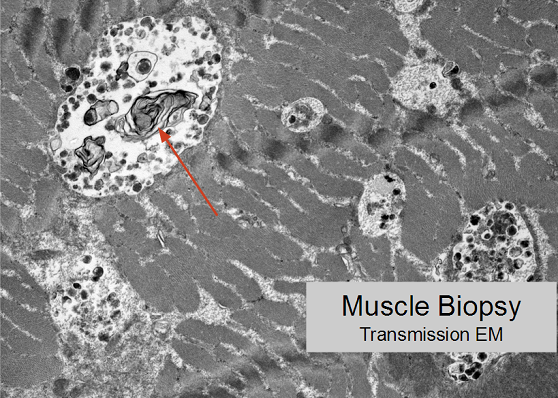
Lysosomal dysfunction develops, characterized by lamellar structures on transmission electron microscopy.
Diagnosis is based on measurement of acid alpha-glucosidase activity within blood. Abnormally low (or absent) acid alpha-glucosidase activity is suggestive of Pompe’s disease.
Treatment for Pompe disease
Treatment consists of supplying acid alpha-glucosidase IV. The protein is taken up by lysosomes, reconstituting the lysosomal glycogenolysis function.
There are two forms of the therapy, both of which are alglucosidase alfa. Myozyme is used for infantile acid maltase deficiency, and has been shown to increase ventilator-free survival (Nicolino et al., 2009). The earlier the diagnosis and treatment, the better the outcomes.
For late-onset acid maltase deficiency Lumizyme has been shown to improve functional capacity, including walking distance and forced vital capacity (van der Ploeg 2010).
The main difference between Lumizyme and Myozyme is the production method, so they are considered separate drugs, are individually approved for the different disorders, and should not be used interchangeably. Therapy with either usually costs over $300,000 a year.
References
Hagemans M L C et al. Clinical manifestation and natural course of late-onset Pompe’s disease in 54 Dutch patients. Brain 2005;128:671-677
Cupler EJ, Berger KI, Leshner RT, Wolfe GI, Han JJ, Barohn RJ, Kissel JT; AANEM Consensus Committee on Late-onset Pompe Disease. Consensus treatment recommendations for late-onset Pompe disease. Muscle Nerve. 2012 Mar;45(3):319-33. doi: 10.1002/mus.22329. Epub 2011 Dec 15.
Marc Nicolino et al. Clinical outcomes after long-term treatment with alglucosidase alfa in infants and children with advanced Pompe disease. Genetics in Medicine (2009) 11, 210–219.
van der Ploeg AT, Clemens P, Corzo D, Escolar D, Florence J, Groeneveld G, et al. A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N Engl J Med. 2010;362:1396–1406.