Carnitine palmitoyltransferase II (CPT) deficiency is the most common inherited lipid-metabolism disorder in skeletal muscle. The adult form causes exercise-induced myalgias, myoglobinuria (myoglobin excretion into the urine) with rhabdomyolysis and myopathy. Triggers include fever, illness, and a high-fat dietary load. In severe cases with complete loss of enzyme function, the disease is fatal in infancy. When some enzyme activity is retained, strength can be maintained between episodes but renal dysfunction can develop secondary to rhabdomyolysis.
Laboratory findings include low serum carnitine levels (both total and free), elevated CK (especially after exercise episodes), and myoglobinuria.
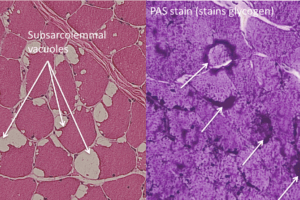
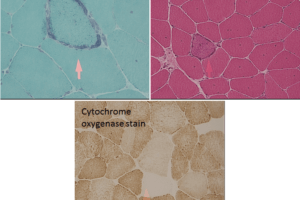
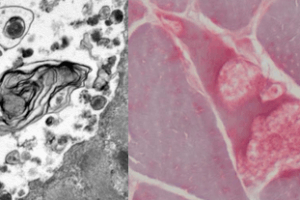
CPT II deficiency has an autosomal recessive inheritance pattern. Unlike mitochondrial myopathy, the abnormal gene causing CPT deficiency is in the somatic genome. Muscle biopsy shows vacuoles on H&E stain and increased granular staining at the periphery of muscle fibers on trichrome stain. Most telling, however, is a positive Red-oil-O stain (staining for lipids) as shown below.

The Muscular Dystrophy Association provides important patient advocacy for metabolic myopathies like this one.
Images courtesy of Dr. Lei Zhao, Dr. Jerry Wong, and Dr. Peter Pytel at the University of Chicago.