McArdle disease is a glycogen storage disease in which the enzyme muscle myophosphorylase is deficient. It is commonly known as glycogen storage disease V. The disorder is characterized by exercise-induced muscle pain, myoglobinuria and elevated serum CK sometimes to the degree of clinical rhabdomyolysis with renal dysfunction. Patients have a “second-wind” phenomenon once non-glycogen dependent energy stores can be engaged. This typically happens about 10 minutes after starting aerobic exercise.
There is usually no family history since the disorder is autosomal recessive. The disorder is rare, with estimates of prevalence ranging between 1:100,000 and 1:200,000.
Differential diagnosis for McArdle Disease
The differential includes other glycogen storage diseases, a variety of somatic genetic myopathies, idiopathic disorders (like toe-walking), mitochondrial myopathy disorders, and other causes of myoglobinuria or rhabdomyolysis. Disorders like CPT deficiency have some similarities, although triggers and pathology differ. The decision to assess different causes depends on the presenting symptoms, age of onset, and severity.
Pathological findings in Glycogen Storage Disease V
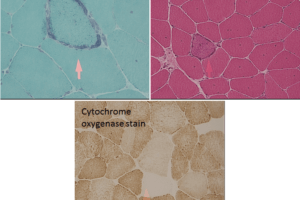
Classic pathological findings on muscle biopsy are shown in the images below, and include:
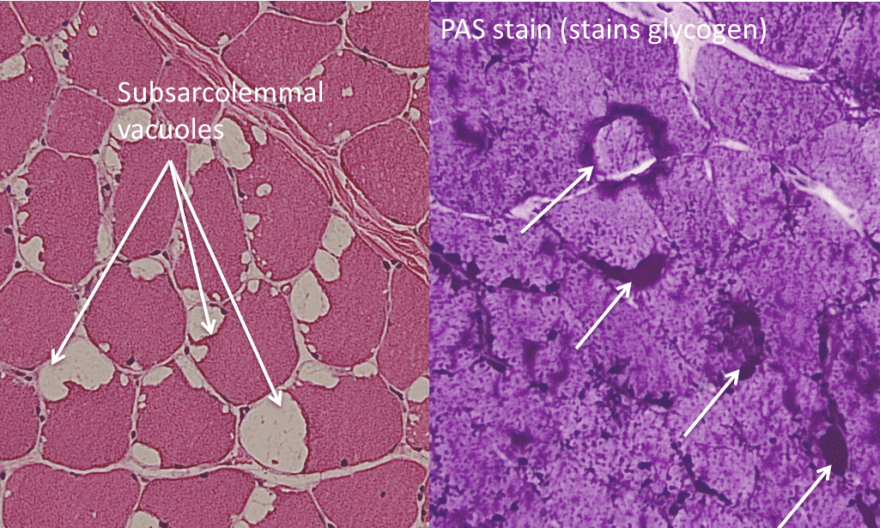
- Sub-sarcolemmal vacuoles that are dark on PAS stain for glycogen.
- Absent myophosphorylase staining.

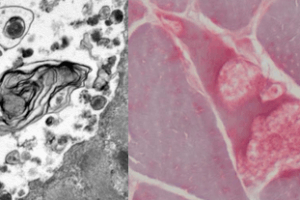
 McArdle Disease shows absent myophosphorylase staining; inset shows myophosphorylase staining in a normal control.
McArdle Disease shows absent myophosphorylase staining; inset shows myophosphorylase staining in a normal control.Definitive diagnosis can be made by genetic testing. A finding of biallelic muscle form of glycogen phosphorylase (PYGM) with pathogenic variants. Alternatively, enzymatic testing showing absent myophosphorylase strongly supports the diagnosis of McArdle Disease.
Images courtesy of Dr. Lei Zhao, Dr. Jerry Wong, and Dr. Peter Pytel at the University of Chicago.
References
Martin et al., Glycogen Storage Disease Type V. Gene Reviews.
Pomarino et al., McArdle’s disease: A differential diagnosis of idiopathic toe walking. Journal of Orthopaedics. 2018 15:685-689