Creutzfeld-Jacob disease is a rapidly progressive spongiform degeneration of the brain due to the accumulation of misfolded prion proteins (PRNP).
The human prion diseases that have been described include:
- Creutzfeld-Jacob disease (CJD)
- Variant Creutzfeld-Jacob disease (vCJD)
- Kuru (New Guinea)
- Gerstmann-Straussler-Scheinker disease (GSS)
- Fatal Familial Insomnia (FFI)
Several prion diseases occur in animals including bovine spongiform encephalopathy (BSE), scrapie (sheep and goats), and chronic wasting syndrome or (deer spongiform encephalopathy that affects deer and elk). Most experimental studies are done using the mouse prion protein (PrP) in experimental mouse prion disease. Prion disease is transmissible between animals of a species and across species. For example, outbreaks of mad cow disease (BSE) have been attributed to cow-feed containing scrapie-affected sheep brain.
Epidemiology of CJD
CJD is a transmissible dementia but the form of transmission is often unclear, and has an annual incidence of 1 per million people. Most cases are sporadic but 10-15% of cases are familial with autosomal dominant inheritance. Iatrogenic CJD has been seen with infected grafts and stereotactic equipment.
Variant CJD is even more rare and is associated with ingestion of nervous tissue from prion-disease affected animals. There has been concern about the hunting of deer herds affected by chronic wasting syndrome, but there have been no known cases of vCJD linked to affected deer. vCJD has been associated with eating contaminated beef.
Clinical features of CJD
The typical clinical features of CJD include a rapidly progressive dementia, startle myoclonus, and ataxia. The median age of onset for classic CJD is 60 years old with a median survival of less than 6 months. 90% of those affected die within 1 year of symptom onset, although familial cases have a slightly longer clinical course.
There can be subtle prodromal constitutional symptoms that are easily missed including fatigue, malaise, increased sleeping and eating disturbances.
Psychiatric and behavioral disturbances are significant in the early stages in about a third of patients. These include personality changes, anxiety, psychosis, paranoia, depression, obsessive compulsive disorder, or anorexia nervosa.
Motor signs are prominent and can include pyramidal signs, cerebellar ataxia, and extrapyramidal symptoms including rigidity, tremors and choreoathetosis.
Patients may present generalized or partial seizures.
Less common signs and symptoms are oculomotor disorders, paresthesias and vegetative dysfunction. The Heidenhain variant has early visual disturbances and is reported in 20% of CJD cases.
Diagnostic tests for CJD
Brain biopsy and autopsy are the only definitive proof of CJD. Because there is no effective intervention and because it is difficult to disinfect surgical equipment contaminated with prion disease, surgical biopsy to confirm a diagnosis of CJD has fallen out of favor. The case of 8-15 people possible infected by prion protein-contaminated surgical equipment in the US Northeast highlights the risks of surgery in CJD patients.
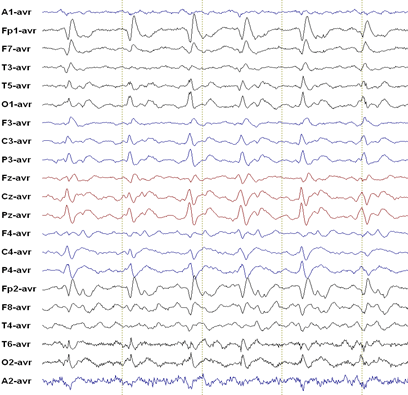
EEG: 70% of patients with CJD have an abnormal EEG, although serial recordings may be required before abnormalities develop. Typical EEG findings of CJD include periodic sharp wave complexes (PSWC) which may disappear as the disease progresses. PSWC may also be preceded by frontal intermittent rhythmic delta activity (FIRDA; to be distinguished from the benign EEG variant SREDA). The classic description of PSWC is a 1 Hz spike and wave complex, but epileptologists note that in reality the EEG of CJD rarely has this “classic” appearance.
CSF shows pleocytosis and mild protein elevation. 14-3-3 Protein in the CSF has a high sensitivity for CJD, but lower specificity.
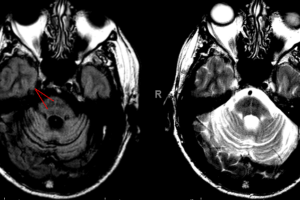
Imaging: Cortical ribboning in DWI sequences is the most common finding on neuroimaging; but this finding is nonspecific and imaging is often more helpful in ruling out other neuropathology.

Neuropathology of CJD
The hallmarks of prion disease are:
- Spongiform degeneration of neurons
- Severe astrocytic gliosis out of proportion to neuron loss
- Amyloid plaque formation
- Lack of inflammatory reaction
Classic CJD displays the most prominent spongiform changes in the regions most commonly seen affected on DWI imaging, including cerebral cortex, cerebellar molecular layer, the putamen and caudate, and the thalamus. The Heidenhain variant shows these changes prominently in the occipital cortex.
New Variant CJD
New variant CJD was first described in 1996 in two teenagers from the United Kingdom. nvCJD occurs in younger patients, has a longer course and has kuru-type plaques in the brain. The median age of onset is 27 years and median survival is more than 1 year. There are prominent psychiatric symptoms of depression and psychosis combined with paresthesias. There is apathy, social withdrawal and sensory symptoms followed by ataxia, pyramidal and extrapyramidal signs and primitive reflexes.
Clinical course of CJD
Rapid progression differentiates CJD from most other mid- to late life dementias. There is no treatment available for CJD, and therapeutic intervention focuses on symptomatic relief for anxiety and neuropsychiatric symptoms.