Wilson disease is an autosomal recessive disorder of copper metabolism. Patients with the disease experience excessive deposition of copper in the liver, brain, and other tissues. It appears that the primary defect is excessive absorption of copper from the small intestine and decreased excretion of copper by the liver. The processes of incorporation of copper into ceruloplasmin and excretion of excess copper into bile are impaired. The copper is stored in the liver, causing hepatocellular damage. Eventually, excess copper is released into the circulation and is deposited into other organs.
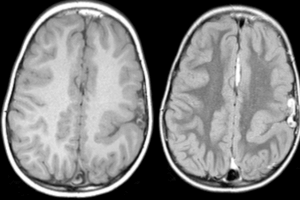
It is a rare disease, having an incidence rate of roughly one in 10 to 30 million cases. The carrier state is 1 per 100 people. Wilson’s disease may take on the form of a fulminant course. When this occurs, there can be progressive liver failure, encephalopathy, coagulopathy, and death if emergent liver transplantation is not performed.
Like most metabolic diseases, Wilson’s disease can have a variety of clinical presentations.
Liver disease of an unexplained etiology in a patient younger than 40 should raise it as a possibility. It may, however, present as an acute hepatitis.
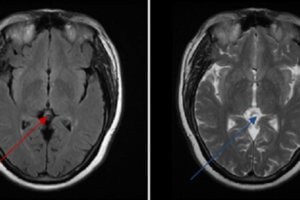
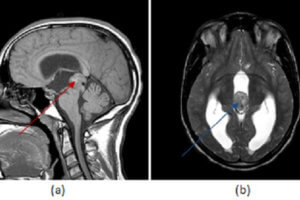
Usually, patients with Wilson’s disease who have neuropsychiatric symptoms also have cirrhosis. The most common symptom is a tremor, but difficulty speaking, excessive salivation, ataxia, masklike facies, clumsiness with the hands, and personality changes may all be early signs of the disease. Psychiatric features include emotional lability, impulsiveness, disinhibition, and self-injurious behavior.
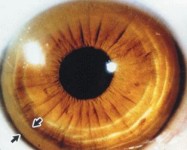
The most common ophthalmologic finding is Kayser-Fleischer rings, formed by the deposition of copper in the Descemet membrane in the limbus of the cornea.
These appear in up to 90% of individuals with symptomatic Wilson disease—almost always in those with neurologic manifestations. They may also be seen in patients with chronic cholestatic disorders, such as partial biliary atresia, primary biliary cirrhosis, primary sclerosing cholangitis, and cryptogenic cirrhosis and should not be considered as exclusive to Wilson’s disease.
Patients with Wilson’s disease may also have skeletal involvement. Over half have osteopenia on x-rays. 20% to 50% of patients have joint disease. It is usually a late manifestation.
Renal involvement may include a syndrome that resembles Fanconi syndrome or urolithiasis because of hypercalcemia.
Diagnosis of Wilson’s disease is completed through an assessment of copper metabolism that involve Serum ceruloplasmin, 24 hour measurements of urine copper, serum copper, and hepatic copper.
Oral penicillamine is the treatment for Wilson’s disease. Trientine has been developed as an alternative for those patients who do not tolerate penicillamine.